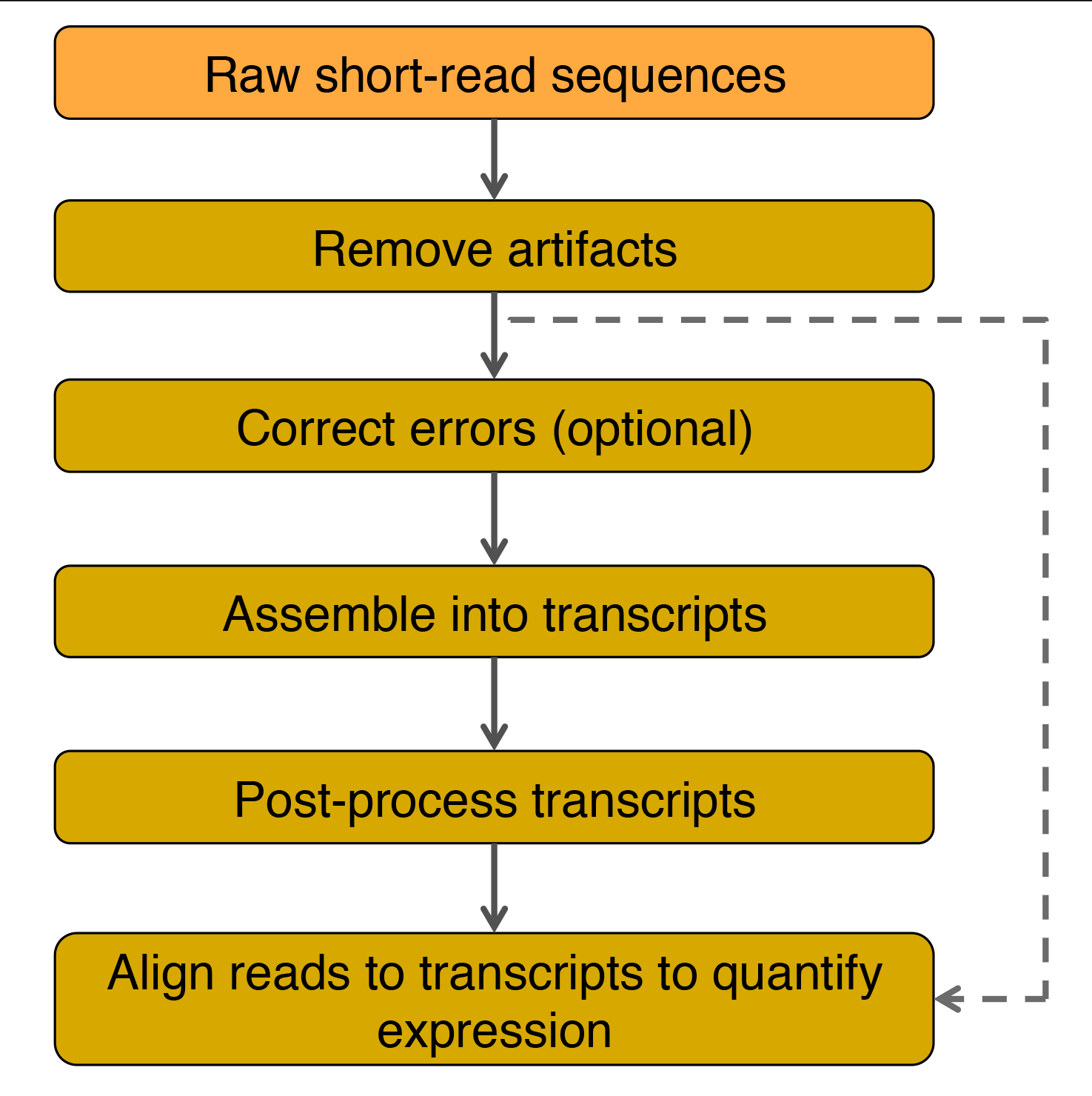
Flow chart for transcriptome assembly and quantification of gene expression. Adapted from Martin and Wang (2011).
Data Analysis
For details see Martin and Wang (2011).
Transcriptome assembly falls into three main categories
- Reference-based strategy: Requires the availability of a reference genome.
- RNA-Seq reads are aligned to a reference genome with a splice-aware aligner.
- Overlapping reads from each locus are clustered to build a graph representing all possible isoforms.
- The graph is traversed to identify individual isoforms.
- De novo strategy: does not require a reference genome; uses de Bruin graph theory (see Compeau et al, 2011) and leverages the redundancy between short read sequences.
- For a set of k-mers from each read, de novo assemblers look for overlapping regions of length k-1 in the k-mers. K-mers are connected if there is exactly a k-1 overlap, and a de Bruijn graph is constructed.
- Connected k-mers are collapsed to form longer sequences until a divergence in the graph is reached based on sequences from a different read. The graph may rejoin or diverge multiple times before all the k-mers re collapsed into a summarizing graph.
- Multiple isoforms are assembled by traversing the collapsed graph.
- Combined strategy: Uses both the reference-based and de novo strategies.
Assembled transcripts are post-processed to remove assembly errors and the expression level of each transcript is estimated by counting the number of reads that align to the transcript