Adapted from Martin and Wang (2011). bp = base pairs
Sample Preparation: Creating a cDNA library
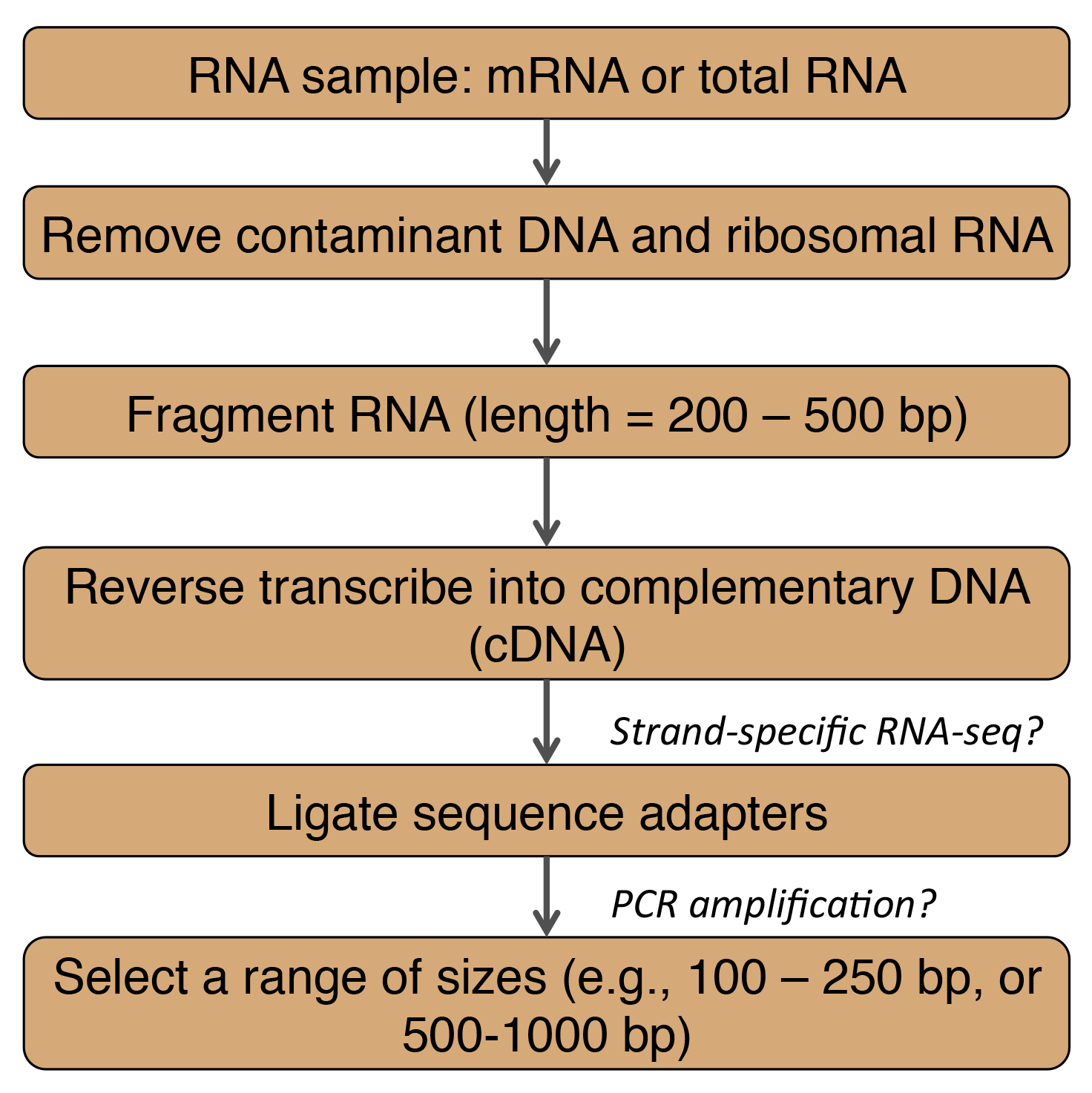
RNA-Seq can be performed on bulk tissue samples or for an individual cell (Martin and Wang, 2011; Mutz et al., 2013; Stegle et al., 2015; Wang et al., 2009; Wilhelm and Landry, 2009). As of 2011, the Helicos system was able to directly sequence RNAs without reverse transcription to cDNA (Martin and Wang, 2011) though other next generation sequencing instruments require a cDNA library. RNA processing includes (see figure):
- Removal of genomic DNA, and ribosomal RNA (rRNA) which is typically >80% of the RNA in cells (Qian et al., 2014).
- For DNA removal use DNase
- For rRNA removal use a depletion kit such as RiboMinus (Invitrogen)
- mRNA usually contains a polyadenylated (polyA) tail. This region can be used to select for mRNA by binding with beads that have long oligo dT stretches (e.g., OligoTex by Qiagen).
- Fragmentation of RNA into smaller lengths that are ~200 to 500 base pairs (bp) by hydrolysis or nebulization. Note the order of this step and the next reverse transcription step can be switched.
- Reverse transciption with random primers or oligo dT primers.
- Strand specificity is important for gene dense genomes (e.g., bacteria, archaea, and lower eukaryotes), and for detection of antisense transcription in higher eukaryotes (Martin and Wang, 2011). For a description of methods see Levin et al. (2010).
- Sequence adapters are added to cDNA fragments to facilitate the amplification the fragments.
- PCR amplification with primers complementary to the ligated adapters may be needed if the instrument sensitivity is not high enough for single molecule sequencing (Qian et al., 2014).
- Ligated cDNA fragments can be separated on 2% Tris-acetate-EDTA agarose gel (Illumina Genome Analyzer II protocol described in Wilhelm et al., 2009). For this protocol, fragment size separation comes before PCR amplification.